Isoform-specific Perturb-seq
Attention
*Under construction*
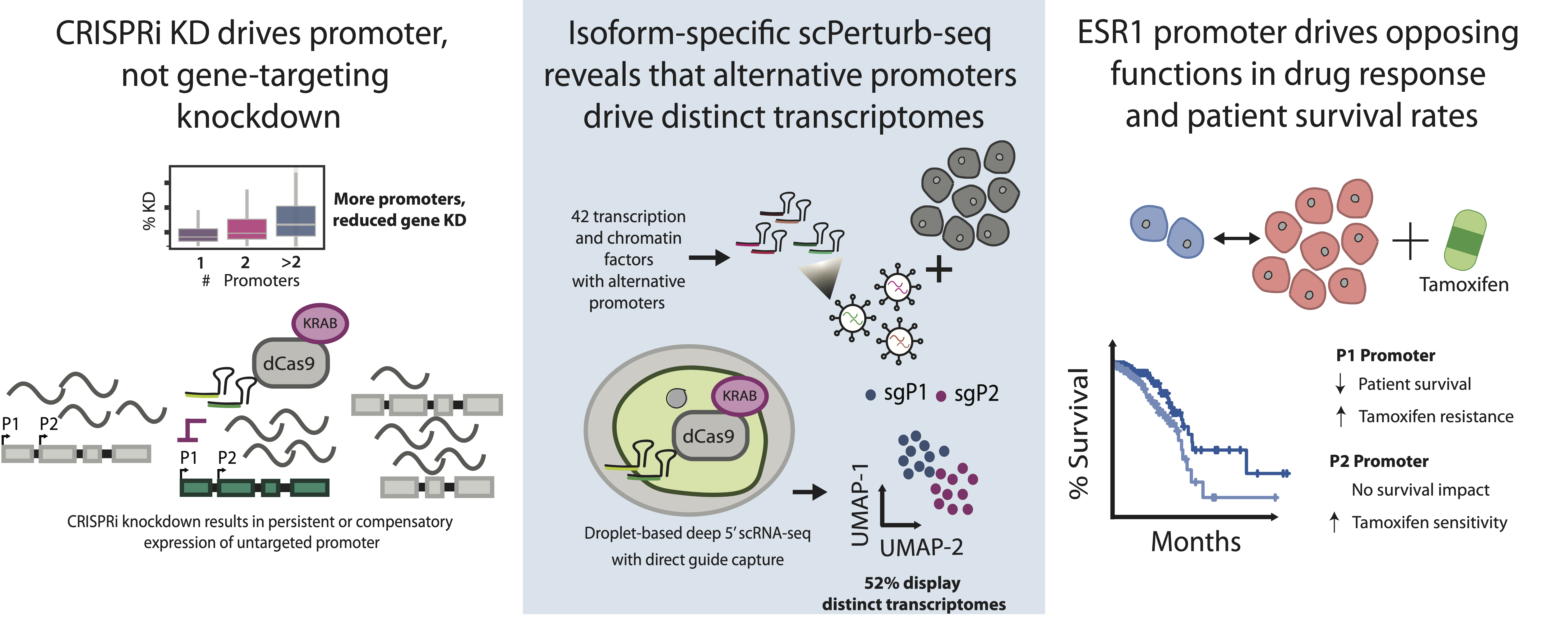
Isoform-specific single-cell Perturb-seq reveals that alternative promoters (AP) are not just redundant regulatory elements but drive distinct biological programs. This documentation covers the methods used to identify these promoters and quantify their functional impact on gene regulation and drug response.
Abstract and Key Findings
CRISPR-dCas9 technologies are typically designed to modulate gene expression at the gene level. However, many hits are promoter and isoform-specific. By leveraging the spatial specificity of CRISPRi—which typically influences transcription within ~1000 nucleotides of the guide binding site—we developed an isoform-specific Perturb-Seq screen.
Major Biological Insights:
Widespread Functional Divergence: Alternative promoters drive functionally distinct biological programs in 51.6% of surveyed genes.
Limited Transcriptional Overlap: Promoter-specific knockdowns (P1 vs P2) typically perturb dozens to hundreds of genes, but only a small minority (median ~10) overlap between the two promoters of the same gene.
Cell Cycle Regulation: Alternative promoters frequently control divergent pathways in cell cycle regulation and proliferation.
Functional Case Study: ESR1 and Tamoxifen Response
The Estrogen Receptor 1 (ESR1) serves as a primary example of how alternative promoters influence clinical outcomes and drug sensitivity in breast cancer.
Isoform Structure: Coordinated splicing between the P1 promoter and an alternative last exon produces a protein isoform with differences in the AF2 domain, potentially modifying interactions with estrogen and selective estrogen receptor modulators (SERMs).
Clinical Significance: High expression of the P1 promoter strongly correlates with decreased survival in Luminal-A breast cancer patients (HR = 1.9), while P2 expression shows no such association.
- Drug Response:
P2 Knockdown: Increases sensitivity to tamoxifen and significantly reduces cellular proliferation.
P1 Knockdown: Leads to increased proliferation in the presence of tamoxifen, suggesting a role in drug resistance.
Analysis Pipeline Overview
The CRISPRi pipeline uses dual guide perturbation [1]. This analysis is structured into three main phases:
Promoter Identification Integration of RNA-seq [2] processed with proActiv [3], ChIP-seq [4], long-read RNA-seq [5], and CAGE-seq [6] to identify targetable distal alternative promoters missed by standard CRISPRi libraries.
Guide Design Utilizing FlashFry [7] and CRISPR-DO [8] for promoter-specific dual-guide design to ensure spatial targeting within the window of CRISPRi effectiveness.
APU Perturb-seq Analysis Quantifying functional divergence through:
Transcriptomics: Using Whippet [9] for isoform-specific quantification post-UMI deduplication.
Pathway Activity: Using Spectra [10] to identify coordinated gene expression programs associated with specific promoters.
Chromosomal Dynamics: Using inferCNV [11] to identify increases in copy number variations.
Cell Cycle Modeling: Using Mahdessian et al. [12] and Seurat [13] to model cell cycle phase distributions across promoter knockdowns.
Clinical Correlations: Using GEPIA2 [14] to link promoter-specific expression patterns to patient survival data.
References
Replogle, J.M., Norman, T.M., Xu, A., Hussmann, J.A., Chen, J., Cogan, J.Z., Meer, E.J., Terry, J.M., Riordan, D.P., Srinivas, N. et al. (2020) Combinatorial single-cell CRISPR screens by direct guide RNA capture and targeted sequencing. Nat Biotechnol, 38, 954-961.
Zhang, J., Lee, D., Dhiman, V., Jiang, P., Xu, J., McGillivray, P., Yang, H., Liu, J., Meyerson, W., Clarke, D. et al. (2020) An integrative ENCODE resource for cancer genomics. Nat Commun, 11, 3696.
Demircioglu, D., Cukuroglu, E., Kindermans, M., Nandi, T., Calabrese, C., Fonseca, N.A., Kahles, A., Lehmann, K.V., Stegle, O., Brazma, A. et al. (2019) A Pan-cancer Transcriptome Analysis Reveals Pervasive Regulation through Alternative Promoters. Cell, 178, 1465-1477 e1417.
Consortium, E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57-74.
Anvar, S.Y., Allard, G., Tseng, E., Sheynkman, G.M., de Klerk, E., Vermaat, M., Yin, R.H., Johansson, H.E., Ariyurek, Y., den Dunnen, J.T. et al. (2018) Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol, 19, 46.
Lizio, M., Abugessaisa, I., Noguchi, S., Kondo, A., Hasegawa, A., Hon, C.C., de Hoon, M., Severin, J., Oki, S., Hayashizaki, Y. et al. (2019) Update of the FANTOM web resource: expansion to provide additional transcriptome atlases. Nucleic Acids Res, 47, D752-D758.
McKenna, A. and Shendure, J. (2018) FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol, 16, 74.
Ma, J., Koster, J., Qin, Q., Hu, S., Li, W., Chen, C., Cao, Q., Wang, J., Mei, S., Liu, Q. et al. (2016) CRISPR-DO for genome-wide CRISPR design and optimization. Bioinformatics, 32, 3336-3338.
Sterne-Weiler, T., Weatheritt, R.J., Best, A.J., Ha, K.C.H. and Blencowe, B.J. (2018) Efficient and Accurate Quantitative Profiling of Alternative Splicing Patterns of Any Complexity on a Laptop. Mol Cell, 72, 187-200 e186.
Kunes, R.Z., Walle, T., Land, M., Nawy, T. and Pe’er, D. (2024) Supervised discovery of interpretable gene programs from single-cell data. Nat Biotechnol, 42, 1084-1095.
Puram, S.V., Tirosh, I., Parikh, A.S., Patel, A.P., Yizhak, K., Gillespie, S., Rodman, C., Luo, C.L., Mroz, E.A., Emerick, K.S. et al. (2017) Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell, 171, 1611-1624 e1624.
Mahdessian, D., Cesnik, A.J., Gnann, C., Danielsson, F., Stenstrom, L., Arif, M., Zhang, C., Le, T., Johansson, F., Schutten, R. et al. (2021) Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature, 590, 649-654.
Hao, Y., Stuart, T., Kowalski, M.H., Choudhary, S., Hoffman, P., Hartman, A., Srivastava, A., Molla, G., Madad, S., Fernandez-Granda, C. et al. (2024) Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat Biotechnol, 42, 293-304.
Tang, Z., Kang, B., Li, C., Chen, T. and Zhang, Z. (2019) GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res, 47, W556-W560.
Analysis Pipeline: